Chronic Myeloid Leukemia (CML)
I. Introduction & Epidemiology
- Chronic myeloid leukemia is a myeloproliferative neoplasm characterized by uncontrolled proliferation of granulocytes (neutrophils, eosinophils, basophils); thrombocytosis may also occur
- Driven by the BCR::ABL1 fusion gene resulting from t(9;22)(q34;q11) — the Philadelphia chromosome.
- Constitutes 15–20% of adult leukemias; incidence 1–2 per 100,000/year
- Median age: 64 years; male predominance (M:F = 1.3:1); rare in children
- Only proven risk factor: ionizing radiation. No other risk factors are known
- Historical note: CML was the first malignancy treated with targeted therapy (imatinib), transforming it from a fatal disease to a chronic condition with near-normal life expectancy
II. Pathogenesis (Figure 1)
CML is driven by a single genetic event — the formation of the BCR::ABL1 fusion gene. Understanding this mechanism explains the disease biology, clinical features, and treatment rationale.
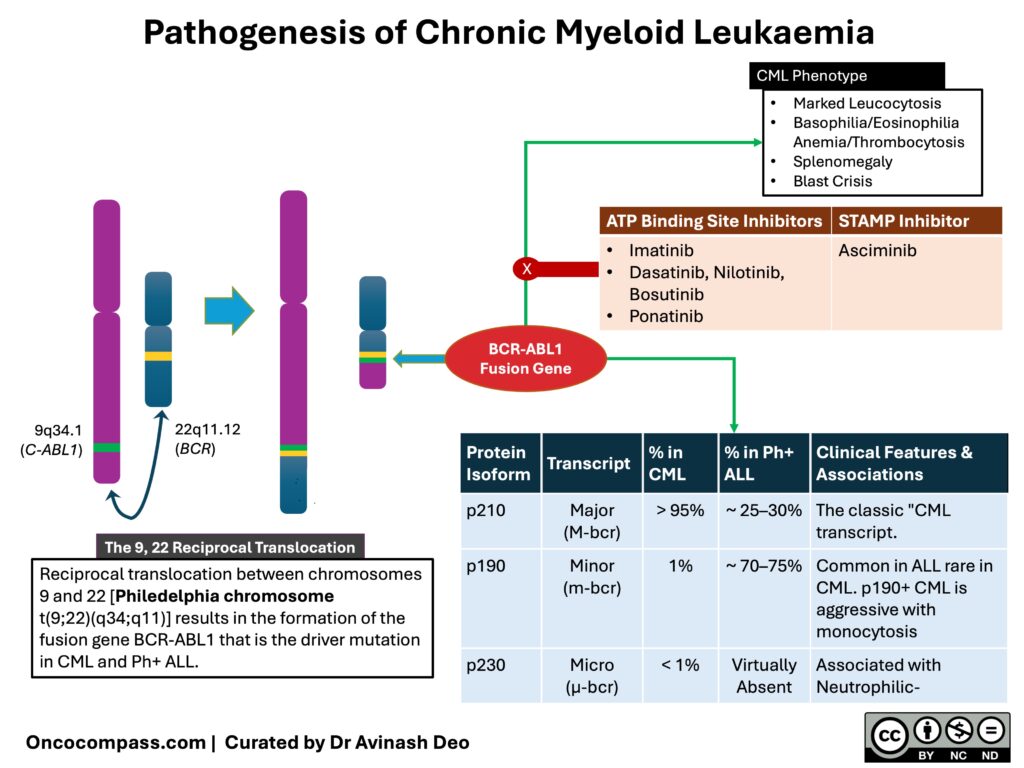
Inhibition of BCR-ABL1 suppresses the leukaemic clone, reverses the features of CML, and restores normal haematopoiesis. The first class of TKIs was the ATP site inhibitors. Imatinib was the first tyrosine kinase inhibitor targeting BCR-ABL1; the second-generation TKIs are more potent and induce a faster and deeper remission. The third-generation TKIs target the T315I mutation, a mutation that confers resistance to other TKIs. The STAMP inhibitors are allosteric inhibitors of BCR-ABL1. (Click here for a sharable PDF of the image)
A. The Philadelphia Chromosome
- A reciprocal translocation, t(9;22)(q34;q11), between chromosomes 9 and 22 creates a shortened chromosome 22 — the Philadelphia (Ph) chromosome
- This fuses the BCR gene (chromosome 22) with the ABL1 gene (chromosome 9), producing the BCR::ABL1 fusion gene
- The fusion gene encodes a constitutively active tyrosine kinase which is the sole driver of CML. It has been associated with uncontrolled granulocyte proliferation, decreased apoptosis, genomic instability, and adhesion defects. This results in leukocytosis with release of immature cells into the blood (chronic phase features), while progressive genomic instability drives accumulation of additional mutations and eventual transformation to blast crisis.
- This “single driver” biology has a critical therapeutic implication: blocking BCR::ABL1 is sufficient to control the disease.
B. Translocation Variants
The classic t(9;22) is present in >90% of cases, but variant forms exist. In
- Complex (variant) translocations (~5%), one or more additional chromosomes participate in the rearrangement alongside chromosomes 9 and 22; the BCR::ABL1 fusion gene still forms and the Philadelphia chromosome is still produced, but the reciprocal exchange involves more than two chromosomes. These are detectable by karyotyping.
- Cryptic translocations (rare), the BCR::ABL1 fusion gene forms through a submicroscopic rearrangement that is invisible on conventional karyotyping — chromosomes appear normal. These require FISH or RT-PCR for detection. The clinical implication is important: a normal karyotype does not exclude CML. If CML is suspected clinically, molecular testing (FISH or RT-PCR) must be performed regardless of karyotype findings.
Table 1. BCR::ABL1 Translocation and Variants
| Type | Frequency | Detection | Clinical Implication |
|---|---|---|---|
| Classic t(9;22) | >90% | Karyotype | Standard |
| Variant (involves 3rd chromosome) | ~5% | Karyotype | Behaves identically; same treatment |
| Cryptic | Rare | FISH/RT-PCR only | Normal karyotype; BCR::ABL1 fusion gene formed |
C. BCR::ABL1 Isoforms
The breakpoint location determines the protein isoform which in turn determines the clinical picture:
Table 2. BCR::ABL1 Isoforms
| Isoform | Transcript | % in CML | Clinical Features |
|---|---|---|---|
| p210 | e13a2 / e14a2 | >95% | Classic CML |
| p190 | e1a2 | ~1% | Monocytosis; aggressive; common in Ph+ ALL |
| p230 | e19a2 | <1% | Indolent; neutrophilic |
Clinical relevance: Identifying the transcript type at diagnosis is essential for molecular monitoring — standard RT-PCR primers detect p210 but may miss p190.
III. Clinical Manifestations & Phases
A. The Natural History of CML
Untreated, CML follows a predictable clinical course through distinct phases:
- Chronic Phase (CP)
- Accelerated Phase (AP)
- Blast Phase (BP)
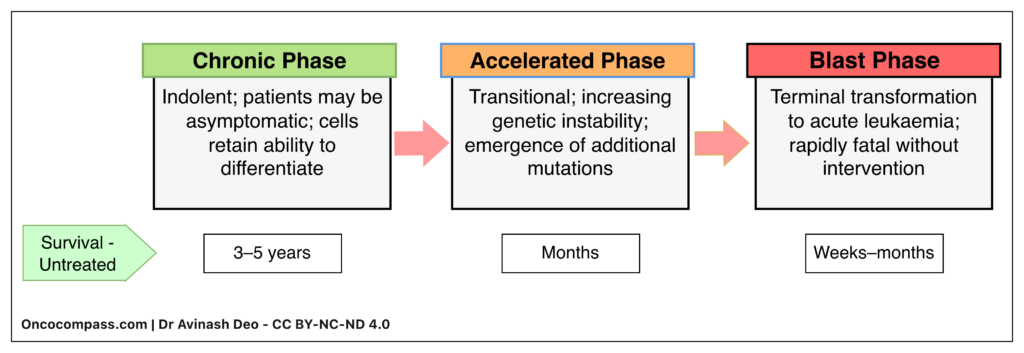
Before the advent of TKIs, median survival was 3–5 years. The patients remained in chronic phase for most of this time. This was followed by a short accelerated phase that culminated in a blast crisis. TKI therapy has transformed this trajectory and converted a uniformly fatal disease into a chronic disease with near-normal life expectancy. TKIs cannot eliminate CML stem cells and cure CML. The goal of therapy is to suppress BCR::ABL1 positive clone and prevent progression to blast phase.
B. Chronic Phase (~85–90% of diagnoses)
The presentation of CML has changed over the years. Asymptomatic patients who are diagnosed on a CBC performed for another reason is now between 20-50%.
Symptoms
- Anaemia-related: Dyspnoea, pallor, palpitations
- Abdominal: Early satiety, left upper quadrant fullness/discomfort (splenomegaly)
- Constitutional: Fatigue, weight loss, night sweats, low-grade fever
- Bone pain / sternal tenderness: From marrow expansion
- Gout: From rapid cell turnover (hyperuricaemia)
Uncommon/Emergency Presentations
- Hyperviscosity/Leukostasis: Extremely high WBC → Increased viscosity and clogging of vessels → Confusion, visual disturbance, respiratory distress, priapism (young males). This is a medical emergency
- Bleeding: Platelet dysfunction despite normal/high platelet count
Signs
- Splenomegaly: Most common and most important finding; often massive; may extend to pelvis
- Hepatomegaly: Less common; usually mild
- Lymphadenopathy: Typically absent — if prominent, consider blast transformation or alternative diagnosis
- Pallor: From anaemia
Clinical Pearl: The combination of massive splenomegaly + leukocytosis + absent lymphadenopathy + basophilia on smear is highly suggestive of CML.
C. Accelerated Phase
A transitional phase between chronic phase and blast crisis, indicating disease progression and decreasing TKI responsiveness.
Suspect AP if any of the following are present:
- Increasing splenomegaly or WBC unresponsive to therapy
- Blasts 10–19% in blood or marrow
- Basophils ≥20% in blood
- Persistent thrombocytopenia or thrombocytosis unrelated to therapy
- New additional chromosomal abnormalities (clonal evolution)
Clinical Note: WHO 2022 removed AP as a formal diagnostic category, but clinical guidelines (NCCN, ELN) retain it for treatment decisions. Students should know both the criteria and the controversy.
D. Blast Phase (Blast Crisis)
Terminal transformation to acute leukaemia — medical emergency with poor prognosis.
Criteria (any one sufficient):
- Blasts ≥20% in blood or bone marrow
- Extramedullary blast proliferation (myeloid sarcoma/chloroma)
- Large clusters/sheets of blasts on marrow biopsy
Clinical presentation:
- Resembles acute leukaemia: bleeding, infections, fever
- Rapidly enlarging spleen
- New or worsening cytopenias
- Bone pain, fatigue, weight loss
Lineage:
- Blast crisis may be lymphoid or myeloid, lymphoid blast crises are almost exclusively B lymphoblastic, T lymphoblastic blast crises are exceptionally rare
- The phenotype of blast crisis in adults and children differs
- Adults: 70% myeloid, 30% lymphoid
- Children: 70% lymphoid, 30% myeloid
Table 4. Phases of CML
| Feature | Chronic Phase | Accelerated Phase | Blast Phase |
|---|---|---|---|
| Blasts (blood/BM) | <10% | 10–19% | ≥20% |
| Basophils | Elevated (universal) | ≥20% | Variable |
| Clinical behaviour | Indolent; often asymptomatic | Intermediate; progressive | Aggressive; acute leukaemia |
| TKI response | Excellent | Reduced | Poor; not durable |
| Prognosis | Near-normal life expectancy | Guarded | Poor (<12 months without HCT) |
| Treatment approach | TKI monotherapy | 2G-TKI; consider HCT evaluation | TKI + chemotherapy; bridge to HCT |
IV. Diagnosis
CML should be considered in any patient with unexplained leukocytosis. While the classic presentation includes splenomegaly and basophilia, many patients are now diagnosed incidentally on routine blood counts — without symptoms or physical findings. CML may also present with isolated thrombocytosis.
A. Diagnostic Workflow
CML usually presents as leucocytosis but rarely may present as isolated thrombocytosis. The diagnostic workup for a suspected case of CML follows a sequential pathway answering six key questions:
- Is there an obvious reactive cause? One must first exclude infections, inflammation, allergies, and drugs.
- Is it Myeloid or Lymphoid? If no reactive cause is found, or if the count is unexplained, the patient is evaluated for neoplasia. The peripheral smear is used to determine the lineage.
- Is it Acute or Chronic? If the lineage is myeloid, does the smear show a “leukemic gap” (Acute) or a full maturation spectrum (Chronic)?
- Is BCR::ABL1 present? This is the confirmatory step for CML.
- What is the Phase? If BCR::ABL1 is positive, the blast percentage determines if the patient is in Chronic, Accelerated, or Blast Phase.
- What is the Risk Category? Finally, the ELTS score is calculated to guide treatment selection.
Note: CML is the only Myeloproliferative Neoplasm (MPN) defined by the presence of BCR::ABL1. If the BCR::ABL1 is negative, the diagnosis is not CML
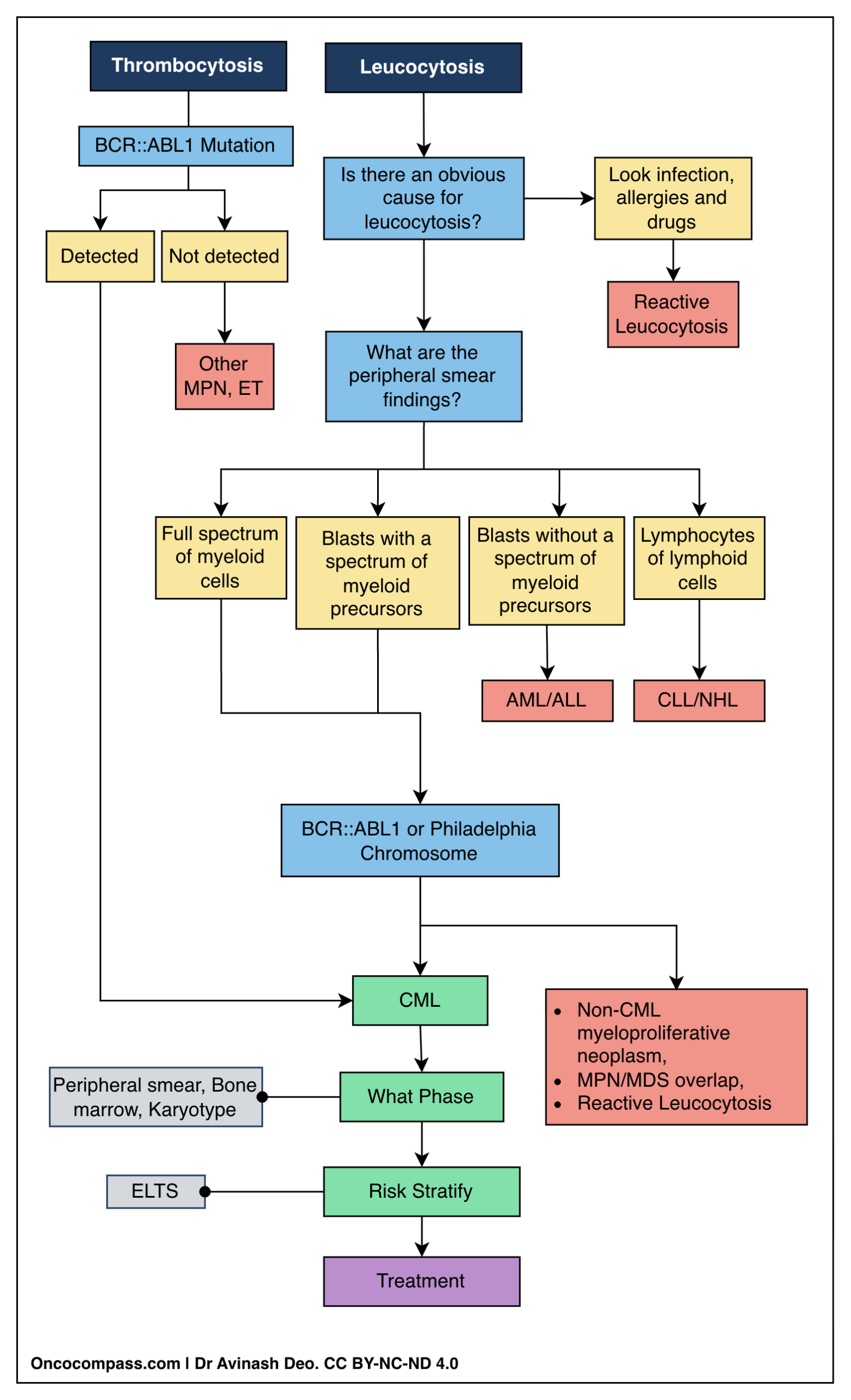
Abbreviations: ET, essential thrombocytosis; ELTS, EUTOS Long-Term Survival score; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; NHL, non-Hodgkin lymphoma.
B. Laboratory Findings In CML
1. Hemogram
- Leucocytosis: WBC often >100,000/µL
- CML: Full spectrum of myeloid maturation (blasts → promyelocytes → myelocytes → metamyelocytes → bands → neutrophils)
- Basophilia is a universal finding in CML, usually it is between 3-5%. This resolves with TKI therapy. If a patient with leukocytosis does not have basophilia a diagnosis other than CML must strongly be considered. Basophilia increases with progression. Accelerated phase is diagnosed if there are > 20% basophils in peripheral blood. Basophilia is responsible for pruritus in CML patients.
- Eosinophilia is seen in 90% of the CML patients.
- Anaemia: Typically moderate normocytic and normochromic that results from bone marrow infiltration and cytokine-mediated suppression. Anaemia becomes more severe with accelerated phase and blast crisis.
- Thrombocytosis: About one third of the patients present with thrombocytosis. Occasionally a patient may present with isolated thrombocytosis, mimicking essential thrombocythemia
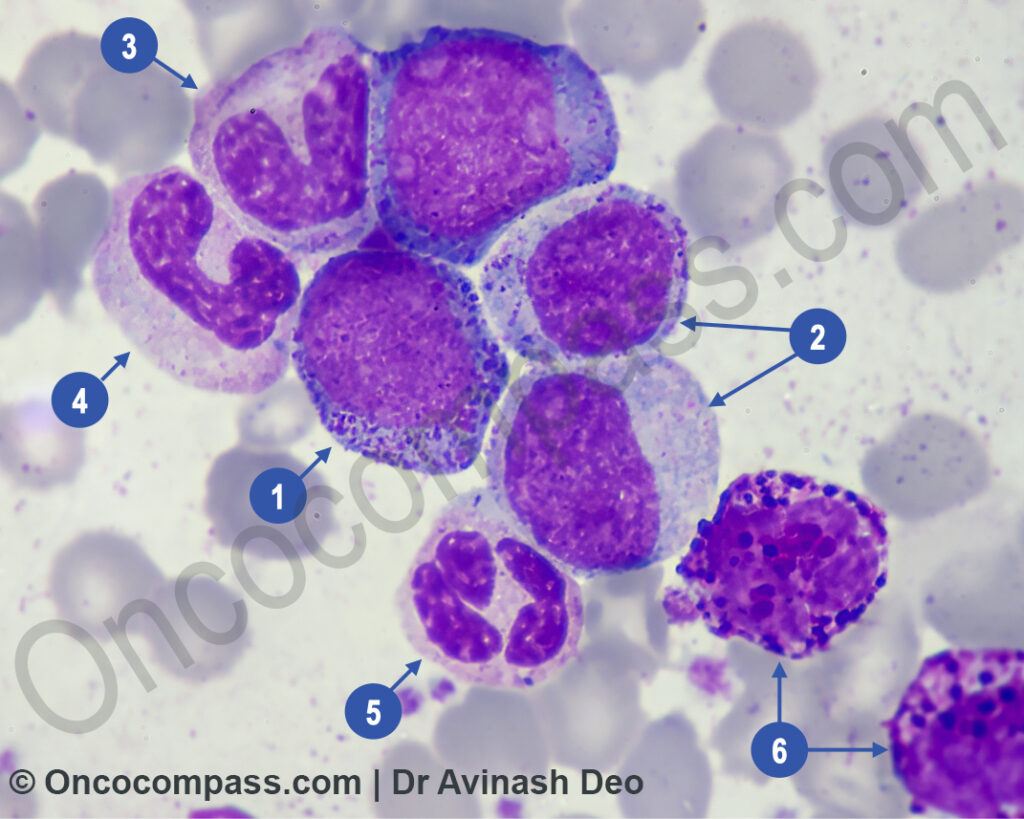
Diagnostic Note: This continuous maturation distinguishes CML from Acute Myeloid Leukemia (AML). In AML, one typically sees a “Leukemic Gap” (historically referred to as Hiatus Leukaemicus)—a predominance of blasts and mature cells, with very few or no intermediate forms (myelocytes/metamyelocytes) in between.
2. Bone Marrow Aspiration & Biopsy
- Marked Hypercellularity with expansion of granulocytic lineage, leading to a significantly increased Myeloid:Erythroid (M:E) ratio (often >10:1 compared to the normal 3:1).
- “Dwarf” Megakaryocytes are a characteristic of CML. These are smaller in size with hypolobated nuclei.
- Blast Percentage
- Chronic Phase: <10% blasts
- Accelerated Phase: 10–19% blasts
- Blast Phase (Blast Crisis): ≥20% blasts (WHO 2022) or ≥30% blasts (older criteria) [See note section IIIC]
- Fibrosis: Reticulin staining is performed to assess for marrow fibrosis (scarring); significant fibrosis is an adverse feature often signaling disease progression.
C. Biochemistry
- Uric Acid: Frequently elevated (Hyperuricemia) due to rapid breakdown of nucleic acids from dying leukemic cells.
- Lactate Dehydrogenase (LDH): Markedly elevated, reflecting the total tumor burden.
D. Karyotype and Molecular Studies
Confirmation of the presence of BCR::ABL1 fusion, that may be done by conventional karyotyping or molecular studies, is the absolute requirement for diagnosis of CML. Karyotyping studies should be performed at diagnosis as they also detect additional chromosomal anomalies some of which are associated with poor prognosis.
1. Conventional Cytogenetics (Karyotype)
- Sample: Required Bone Marrow[preferred] or peripheral blood.
- Results:
- The Philadelphia (Ph) Chromosome [t(9;22)(q34;q11)] is visible in 90–95% of cases.
- Additional Chromosomal Abnormalities (ACAs): This includes High-Risk ACAs: Presence of i(17q), -7/del(7q), or 3q26.2 rearrangements that predict a poorer prognosis and risk of progression.
2. Fluorescence In Situ Hybridization (FISH)
- Sample: Peripheral Blood or Bone Marrow.
- Fluorescent probes to visualize the fusion of BCR and ABL1 genes in interphase (non-dividing) cells.
- Identifies the ~5% of patients who are “Ph-negative” on karyotype but BCR::ABL1 positive (cryptic translocations).
- Excellent for initial diagnosis but generally not sensitive enough for monitoring
3. Quantitative Reverse Transcription PCR (RT-qPCR)
- Sample: Peripheral Blood (Venous blood is sufficient; marrow is not needed).
- The most sensitive test available.
- The International Scale (IS): PCR results vary between labs. To ensure consistency, results are converted to a standardized International Scale (IS).
- Usage: This is the standard tool for lifelong monitoring of treatment response.
4. Kinase Domain Mutation Analysis
- Test: Detection of mutations in the ABL1 kinase domain.
- Indication: Not performed at baseline. Performed only if the patient fails to respond to TKI to detect drug resistance to guide the choice of 2nd-line TKI
V. Differential Diagnosis
The differential diagnosis of CML includes reactive causes, other chronic leukaemias, acute leukaemias, and other myeloproliferative neoplasms.
| Feature | Chronic Myeloid Leukemia (CML) | Acute Leukemia | Leukemoid Reaction (Reactive) |
|---|---|---|---|
| Maturation Pattern | Full Spectrum (Continuous)Shows every stage of differentiation. | "Leukemic Gap" (Hiatus Leukaemicus)Missing the intermediate stages. | Left Shift (Mature Dominance)Shifted towards immature forms but retains mature dominance. |
| Cell Spectrum | All stages present:Blasts → Promyelocytes → Myelocytes → Metamyelocytes → Bands → Neutrophils. | Discontinuous:Predominantly Blasts and Mature Cells. Few or no intermediate myelocytes/metamyelocytes. | Mature forms:Mainly Neutrophils and Bands. Occasional myelocytes/metamyelocytes may be seen. |
| Blasts | Present(Typically <10% in Chronic Phase). | Predominant(Defining feature, >20%). | Absent(Or exceedingly uncommon/rare). |
| Basophilia & Eosinophilia | PresentAbsolute basophilia is a nearly universal finding; eosinophilia is very common. | Absent(Usually not a feature). | Absent(Toxic granulation and Döhle bodies are common instead). |
| Thrombocytosis | Common(30–50% of cases; may be marked). | Absent(Thrombocytopenia is the classic finding). | Possible(Reactive thrombocytosis often accompanies inflammation/infection). |
A. Reactive Leukocytosis (Leukemoid Reaction)
- What it is: Marked leukocytosis (often >50 × 10⁹/L) in response to severe infection, inflammation, or steroid use.
- Why it can be confused with CML: High WBC count with left shift mimics CML.
- Distinguishing features:
- Toxic granulation and Döhle bodies in neutrophils
- High LAP score (low in CML)
- No basophilia
- No splenomegaly
- Resolves when underlying cause is treated
| Condition | Why It Can Be Confused with CML | Key Distinguishing Features |
|---|---|---|
| Reactive leukocytosis (Leukemoid reaction) | High WBC count with left shift mimics CML | Toxic granulation and Döhle bodies; high LAP score; no basophilia; no splenomegaly; resolves with treatment of underlying cause |
| Chronic Lymphocytic Leukaemia (CLL) | Both present with leukocytosis in elderly; often discovered incidentally | Lymphocytosis not granulocytosis; mature small lymphocytes with smudge cells; lymphadenopathy common; no basophilia |
| Acute Leukaemia (AML/ALL) | CML-blast phase mimics de novo acute leukaemia; high WBC in acute leukaemia may be mistaken for CML | ≥20% blasts; leukemic gap (blasts + mature cells; intermediates absent); CML shows full maturation spectrum |
| Other MPNs (PV; ET; PMF) | All are clonal myeloid disorders; may present with splenomegaly and elevated counts | BCR::ABL1 negative; JAK2/CALR/MPL positive; PV: erythrocytosis dominant; ET: thrombocytosis dominant; PMF: marrow fibrosis with leukoerythroblastic picture |
B. Chronic Lymphocytic Leukaemia (CLL)
- What it is: A chronic leukaemia of mature B lymphocytes
- Why it can be confused with CML: Both present with leukocytosis in elderly patients, often discovered incidentally.
- Distinguishing features:
- Lymphocytosis (not granulocytosis)
- Mature small lymphocytes with smudge cells
- Lymphadenopathy common (rare in CML)
- No basophilia
C. Acute Leukemia
- What it is: Malignancy characterised by rapid accumulation of blasts.
- Why it can be confused with CML: CML in blast phase mimics de novo acute leukaemia. Also, acute leukaemia with high WBC may be mistaken for CML.
- Distinguishing features:
- ≥20% blasts
- “Leukemic gap” — blasts and mature cells present; intermediate forms absent
- CML shows full maturation spectrum; acute leukaemia does not
Important: BCR::ABL1 testing should be done in all acute leukaemias — to identify CML-blast crisis and Ph+ ALL, both of which require TKI therapy.
D. Myeloproliferative Neoplasm
- What they are: Polycythaemia Vera (PV), Essential Thrombocythaemia (ET), and Primary Myelofibrosis (PMF).
- Why they can be confused with CML: All are clonal myeloid disorders; may present with splenomegaly and elevated counts.
- Distinguishing features:
- BCR::ABL1 negative
- JAK2/CALR/MPL mutations positive
- PV: erythrocytosis dominant
- ET: thrombocytosis dominant
- PMF: marrow fibrosis, leukoerythroblastic picture
E. Other Rare Conditions
The following rare conditions may also mimic CML but are uncommon and typically encountered at the postgraduate level:
- Chronic Neutrophilic Leukaemia (CNL)
- MDS/MPN with Neutrophilia (formerly Atypical CML)
- Chronic Myelomonocytic Leukaemia (CMML)
- Juvenile Myelomonocytic Leukaemia (JMML)
All are BCR::ABL1 negative.
Key Points
- When in doubt, test for BCR::ABL1.
- BCR::ABL1 is mandatory for CML — if negative, it is not CML
- Basophilia favours CML — absent in reactive causes and most other conditions
- Full maturation spectrum favours CML — leukemic gap suggests acute leukaemia
Reactive leukocytosis, other acute and chronic leukemias, myeloproliferative neoplasm and MPN/MDS overlap may present with leukocytosis.
VI. Treatment of CML Chronic Phase
A. Treatment of a Patient Diagnosed in Chronic Phase
Diagnose → Risk stratify → Screen for comorbidities → Select appropriate TKI → Establish baseline → Initiate therapy promptly → Monitor for response → Modify Therapy as Needed
1. Treatment is Mandatory
- All patients require treatment at diagnosis — no role for “watch and wait”
- Goal: Prevent progression to blast crisis that has high mortality.
- All patients treated with TKIs.
2. Pre-Treatment Workup
- Confirm BCR::ABL1 and identify transcript type (for monitoring), establish a baseline BCR::ABL1 level for monitoring
- Document spleen size and baseline blood counts
- Screen for HBsAg (risk of reactivation on TKI)
- Baseline ECG (QTc), metabolic panel (glucose, lipids), liver/renal function
3. TKI is the Cornerstone
- Tyrosine kinase inhibitor (TKI) therapy is first-line for all patients with chronic phase CML. Imatinib was the first TKI for CML. Second generation TKIs include dasatinib, nilotinib, and bosutinib. The second generation TKIs give deeper and faster responses. Ponatinib is a third generation TKI that is effective in the T315I mutation. Asciminib has a distinct mechanism from the TKIs mentioned above.
- Hydroxyurea and Leukapheresis may be used to rapidly reduce counts.
- Allogeneic stem cell transplant is reserved for TKI failure/advanced phase.
| Drug | Mechanism of Action | Indication | Dose (Chronic Phase) | Key Side Effects |
|---|---|---|---|---|
| Imatinib (1st Gen) | BCR-ABL1 TKI; also inhibits c-KIT and PDGFR, Does NOT Cover T315I | First-line (low/intermediate risk); all phases | 400 mg OD | Oedema; muscle cramps; nausea; diarrhoea; rash; myelosuppression |
| Dasatinib (2nd Gen) | BCR-ABL1 TKI + SRC kinase inhibitor; does NOT cover T315I | First-line (consider in HIGH-RISK); imatinib-resistant/intolerant; avoid in lung disease | 100 mg OD | Pleural effusion; pulmonary hypertension; myelosuppression; bleeding risk |
| Nilotinib (2nd Gen) | BCR-ABL1 TKI (more potent than imatinib); does NOT cover T315I | First-line (consider in HIGH-RISK); imatinib-resistant/intolerant; avoid in cardiac/metabolic disease | 300 mg BD (first-line); 400 mg BD (resistant) | QT prolongation; hyperglycaemia; dyslipidaemia; pancreatitis; arterial occlusive events |
| Bosutinib (2nd Gen) | BCR-ABL1 TKI + SRC kinase inhibitor; does NOT cover T315I | First-line (consider in HIGH-RISK); resistant/intolerant to prior TKIs; avoid in hepatic impairment | 400 mg OD (first-line); 500 mg OD (resistant) | Diarrhoea (prominent); nausea; vomiting; hepatotoxicity; rash |
| Ponatinib (3rd Gen) | BCR-ABL1 TKI; COVERS T315I mutation | T315I mutation; resistant/intolerant to ≥2 TKIs | 45 mg OD (reduce after response) | Arterial thrombosis (serious); hypertension; pancreatitis; hepatotoxicity |
| Asciminib (STAMP inhibitor) | Allosteric inhibitor (binds ABL myristoyl pocket); COVERS T315I | Resistant/intolerant to ≥2 TKIs; T315I mutation | 40 mg BD (general); 200 mg BD (T315I) | Myelosuppression; musculoskeletal pain; hypertension; fatigue; pancreatitis |
4. Risk Stratify Before Initiating Treatment
- Calculate risk score at diagnosis (ELTS preferred)
- Low/Intermediate risk: Imatinib is appropriate (cost-effective, well-tolerated, long-term safety data)
- High risk: Consider 2nd generation TKI upfront (dasatinib, nilotinib, or bosutinib) for faster, deeper responses and presumably reduced progression risk
5. Co-morbidities Influence Choice
- Lung disease / pleural effusion risk → Avoid dasatinib
- Cardiac disease / QT prolongation / diabetes / dyslipidaemia → Avoid nilotinib
- Hepatic impairment → Avoid bosutinib
- No significant comorbidities → Any TKI appropriate; consider cost and monitoring requirements
6. Patient Counselling
- Lifelong therapy (in most cases, see TFR below) — adherence is critical
- TKI therapy should be avoided during pregnancy (teratogenic, especially 1st trimester)
- Discuss contraception. Emphasize the risks of an unplanned pregnancy.
- Discuss side effect profile specific to chosen TKI
E. Monitoring of Therapy
The aim of regular monitoring is to detect loss of response or progression early. Patients should be monitored every three months. A CBC and BCR::ABL1 by RT-PCR(IS) should be done.
| Test | Detects | Primary Role | Frequency |
|---|---|---|---|
| CBC | Cell counts, differentials | hematological response, toxicity | every 2 weeks till stable then once in 3 months |
| RT-PCR (IS) | BCR::ABL1 transcript level | Molecular response; early relapse | Every 3 months, frequency may be reduced to once in 6 months in patinets who have a deep response |
| Karyotype | Ph chromosome; ACAs | Cytogenetic response; clonal evolution | At diagnosis to determine stage, document baseline ACAs; at failure to detect ACA |
| Mutation analysis | ABL1 kinase domain mutations | TKI resistance; guides drug switch | At treatment failure only, to decide about the TKI that will be effective. |
Depending on the degree of suppression of BCR::ABL1 four types of responses are defined.
- Complete Haematological Response (CHR): No clinical or hematological evidence of disease is usually achieved in 3 months
- Complete Cytogenetic Response (CCyR): CCyR is defined as BCR::ABL1 < 1%. It is usually achieved in 3-6 months
- Major molecular response (MMR): MMR is defined as BCR::ABL1 ≤0.1% (IS) and is usually achieved by 9-18 months. MMR is achieved faster with 2nd generation TKIs and asciminib. A patient who achieves and maintains MMR has almost no risk of progression to AP or BP
- Deep Molecular Response (DMR): DMR may be MR4 (≤0.01%) or MR4.5 (≤0.0032%). It takes years to achieve and is necessary for considering discontinuation of therapy (see TFR)
Patients who have a slow fall in BCR-ABL1 transcript have a higher chance of recurrence. These patients must be assessed for compliance and if compliant then change in TKI should be considered.
VII. Management of Advanced Phases
A. Accelerated Phase (AP)
There are two types of APs de novo AP and transformed AP that carry a different prognosis and need a different approach
1. De Novo AP (Diagnosed at Presentation)
- Patients who present with features of Accelerated Phase at the time of their initial diagnosis, without prior treatment.
- Aggressive disease, not exposed to drugs, likely to be drug sensitive
- Patients have a Better prognosis than those with transformed AP and can achieve long term control on TKI.
- They should be treated with Second-Generation TKIs
- Therapy should not be discontinued even if they achieve a sustained deep molecular remission (see treatment free remission below).
2. Transformed AP (Progression on Therapy)
- Patients who were initially diagnosed in Chronic Phase but progressed to Accelerated Phase while taking TKI therapy.
- Have resistant disease that has escaped TKI inhibition.
- Significantly worse prognosis than de novo AP
- Treated with an alternate TKI followed by allogenic stem cell transplant
B. Blast Phase (BP)
- Blast Phase (BP) represents the terminal transformation of CML into an acute leukaemia.
- The Fundamental Goal: Achieve a second chronic phase (CP2) and proceed to allogeneic HCT
- Treated with Combination therapy (TKI + lineage specific acute leukemia chemotherapy) followed by allogenic stem cell transplant in those who achieve remission.
- Kinase domain mutation testing is mandatory for choosing the right TKI.
VIII. Treatment-Free Remission
- They only suppress the BCR::ABL1 positive clone. TKIs cannot eliminate CML stem cells.
- If TKIs are stopped in patients who have pronounced suppression of BCR::ABL1 almost half will remain free of relapse. This is called treatment free remission (TFR)
- Most of the relapses take place within 6 months. Reinitiation of TKIs can suppress the BCR::ABL1 positive clone. Relapses do not hamper survival
- Discontinuation of TKIs spare the patient of long term toxicities and may be considered in patient who will comply with monitoring and will opt for discontinuation.
- Patients who have achieved both the following are eligible for withdrawal of TKIs.
- CML chronic phase only not indicated in AP or BP AND
- Sustained DMR (MR4/MR4.5) ≥2 years AND
- Total TKI duration typically >5 years
- Strict monitoring: Monthly qPCR first 6 months then every 2 between 6-12 months and then every three months indefinitely
- Trigger to restart TKI: Loss of MMR (0.1%)
IX. Special Situations: Pregnancy & Fertility
A. Contraception
- Male Patients
- TKIs do not impair male fertility or increase the risk of miscarriage or fetal abnormalities in female partners of men on TKI
- No TKI discontinuation is required for conception.
- Female Patients
- TKIs are teratogenic and increase the risk of miscarriage. They are contraindicated in pregnancy
- Women on TKI must use contraception.
- Any method may be used but barrier methods may be preferable for patients with cardiovascular risk factors, particularly true for patients taking TKIs that are associated with a cardiovascular toxicity (Nilotinib, Ponatinib)
B. Pregnancy
- Pregnancy should be deferred till TKIs can be discontinued (See TFR above)
- If the conditions for TFR are not met then the patients should be switched to interferon before planning conception
- The washout period for TKI is not known. A month of drugs free period before attempting conception is recommended
- Pre-conception counseling (all patients of childbearing age):
C. Fertility Preservation
- Women: Stop TKI before oocyte retrieval (≥1 month recommended, optimal timing unknown)
- Men: Sperm banking can be performed prior to TKI
D. Breastfeeding
- Contraindicated on TKI (excreted in breast milk)
X. Pediatric CML
- Exceptionally rare; median age at diagnosis: older children
- Higher incidence of p190 (e1a2) transcript (7–10%): aggressive biology, monocytosis
- Blast phase: 70–80% lymphoid (vs. 70% myeloid in adults)
- TKIs first-line (imatinib, dasatinib, nilotinib) — same as adults
- Growth impairment and skeletal maturation delay (especially imatinib in prepubertal children)
- Catch-up growth may occur but many fail to reach expected mid-parental height
- Outcome Comparable to adults with TKI therapy; long-term follow-up ongoing
XI. Prognosis
- Chronic phase: TKIs transformed CML from a fatal disease (median survival 2–3 years) to a chronic condition with 10-year OS >90% and near-normal life expectancy
- Risk stratification: ELTS score is the best predictor of CML-specific mortality
- Advanced disease:
- De novo accelerated phase: 10-year OS ~66%
- Transformed blast phase: median survival ~38 months (poor)
- Modern treatment goal: Beyond survival — achieve deep molecular response to enable treatment-free remission (functional cure without lifelong therapy)
- CP survival: Excellent (PFS ~98%)
- Blast Phase: Poor (median survival <1 year); children slightly better than adults
- TFR: Viable goal; 50–61% successful after sustained DMR
XII. Selected References
- WHO Classification: Khoury JD, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703–1719.
- ELN Recommendations: Hochhaus A, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984.
- NCCN Guidelines: National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology: Chronic Myeloid Leukemia. Version 2.2024. https://www.nccn.org
- CML General Review: Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am J Hematol. 2020;95(6):691–709.
- Treatment-Free Remission Review: Mahon FX, Etienne G. Deep molecular response in chronic myeloid leukemia: the new goal of therapy? Clin Cancer Res. 2014;20(2):310–322.
- Pediatric CML Review: Millot F, Suttorp M, Versluys AB, et al. Chronic myeloid leukemia in children and adolescents. Expert Rev Anticancer Ther. 2017;17(1):21–30.
- CML in Pregnancy Review: Apperley J. CML in pregnancy and childhood. Best Pract Res Clin Haematol. 2009;22(3):455–474.
- Molecular Monitoring Review: Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884.
